This year (2023), multiple papers were published on the cloning of disease-resistance genes in wheat. Three papers described 3 methods for cloning resistance genes without a reference genome: Lr47 cloning with modified MutRNASeq, Lr9/Lr58 cloning with MutIsoSeq, and YrNAM cloning with sequencing trait-associated mutations (STAM). They shared the same procedure:
- Construct a wildtype transcript reference from either short-read RNA-seq data or long-read Isoform sequencing (iso-seq);
- Map the cheap short-read RNA-seq data of the identified susceptible EMS mutants to the reference;
- Variant calling to find genes with mutations in all the mutants. Use the Lr47 cloning process as an example (Li et al. 2023 Fig S6b):
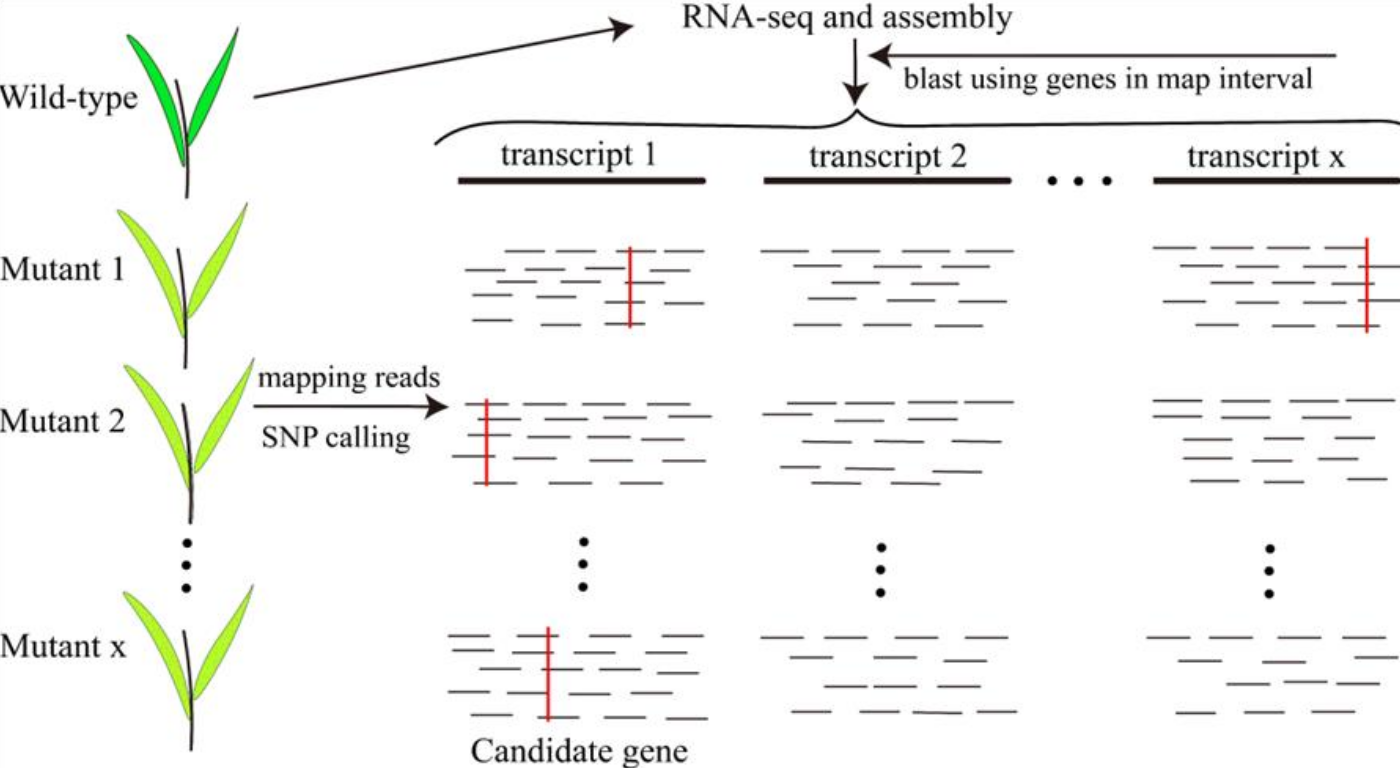
The differences between the 3 methods are as follows:
- Modified MutRNASeq used short-read RNA-seq data to make the wildtype cDNA reference, but MutIsoSeq and STAM used long-read Isoform sequencing (iso-seq). Short-read RNA-seq is cheap but needs assembly, while long-read iso-seq only needs to be cleaned up without assembly.
- MutIsoSeq and STAM are very similar. Based on the YrNAM paper, the main differences are:
- MutIsoSeq keeps multiple Iso-Seq isoforms derived from the same gene, STAM employs a non-redundant full-length transcriptome as a reference.
- MutIsoSeq protocol aims to sequence about 70 Gb RNA-Seq data for each mutant, STAM shows a smaller data set of 15–18 Gb is sufficient. Yr47 cloning only used about 10.5 Gb RNA-seq data, which is also enough, so I think regular RNA-seq (about 10 Gb per sample) is enough for variant calling. You can always sequence more later if your candidate gene has a very low expression level.
Other considerations
These methods are straightforward, but there are other considerations while cloning a resistance gene.
If there are multiple QTLs, select a line with only the QTL you want to clone (other QTLs have the susceptible allele) to do EMS mutagenesis, unless there is only one QTL or the major QTL has much stronger effects (>50% variation explained) than others.
After selecting the candidate gene, design a marker to test the original mapping population to see whether it is co-segregated with the phenotype. I think it is also better to test one wild type x mutant F2 population for co-segregation to make sure that mutation does cause loss of function.
Sequence about 10 susceptible mutants and validate the candidate gene on the remaining mutants without RNA-seq data. All the mutations on the candidate gene could give a map of critical residues required for its function.
Good EMS mutants and solid phenotyping are critical to using these methods.
These methods are good for qualitative traits like disease resistance, but not a good option for quantitative traits like spikelet number per spike. We can still use RNA-seq and its de novo assembly to predict candidate genes based on expression level differences.