In Sanger sequencing, double peaks are common because of the template is not unique sequence, either because your PCR primers are not specific (homeologs in wheat, for example), or the template is from a heterozygous plant, or there are gene duplications. When we send DNAs that might be heterozygous, such as EMS mutants, we will check for double peaks to find the mutation points. Even we think the DNAs are pure, we still need to check for double peaks because of gene duplications or DNA contaminations. The sequencer will call an allele for a double peak, if it is the same as our reference, we may never find it unless we check the chromatogram.
This just happened to me in my experiment. I did not find the double peaks in one of my line’s sequencing data until I found another 2 positions that have different calls from the reference in the reverse primer sequencing. Then I found the gene of this line has a duplication, a mix of two haplotypes. Then I compared the two haplotypes and found there should be another double peak. I checked and there was!
Then I searched and I found the R package “sangerseqR”. It has an awesome introduction manual to help you get familiar with the functions step by step. The most important function of this package is that it can help you find double peaks by making base calls for the primary sequence and the secondary sequence. Then you can produce a graph with highlights of double peaks.
I wrote a R script to produce a chromatogram with double peak highlights for each sanger sequening file (.ab1 or .scf). The usage is easy:
sanger-double-peak.R seq1.ab1 seq2.ab1 seq3.ab1
In the chromatogram, the primary calls (top line) and the secondary calls (2nd line) and the peaks will be presented. Double peaks are highlighed like the figure below:
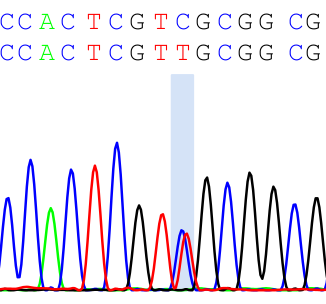
This package can also help you extract primary sequences and secondary sequences. When there are only SNPs, you just need the chromatogram to get the SNP positions. However, if there are indels or insertions, you may want to do a sequence alignment to see where and how big is the indel or insertion. The vignette of this package has a good example of indels.